

成都生物所在高效精准合成糖类衍生物的研究中获进展
时间:2024-04-12
在现代药物研发中,糖类化合物的作用日益受到重视。作为生物大分子之一,糖类化合物在细胞识别、信号传递和免疫反应中扮演着关键角色。然而,由于其复杂的结构和多样的功能,糖类化合物的合成和修饰一直是有机化学和药物化学中的一大挑战。
传统的化学合成方法往往难以实现对糖类化合物结构的精确控制,这限制了其在药物设计中的应用。因此,科学家们一直在寻求更为高效和选择性的合成策略,以便能够在不破坏其活性的前提下,对糖类化合物进行定向修饰。
中国科学院成都生物研究所马小锋课题组在此背景下,一直致力于温和条件下多样化糖苷类化合物的合成方法学与生物活性方面的研究工作(Org. Lett. 2024,26,1332; Molecules 2023,28,4724;Carbohydr. Res. 2023,108902;Chem. Asian J. 2022,17,e202200120; J. Org. Chem. 2022,87,16736)。近期,通过创新的氢键催化策略,成功开发了一种基于硝基烯糖的新型C3官能团化和C1,C3-双官能团化反应方法。这一方法可以在室温条件下,高效、高选择性(包括立体选择性和区域选择性)地合成一系列C1,C3-双吲哚或吡咯取代,以及C1-吲哚-C3吡咯-、C1-吡咯-C3吲哚-取代的糖衍生物,为糖类化合物的合成和修饰提供了新的可能性(图1)。经初步生物活性评价表明,部分化合物具有较强的抗肿瘤活性,并对人体正常肝细胞呈现较低的细胞毒性,具有成为高效、低毒的新型抗肿瘤先导化合物的潜力。
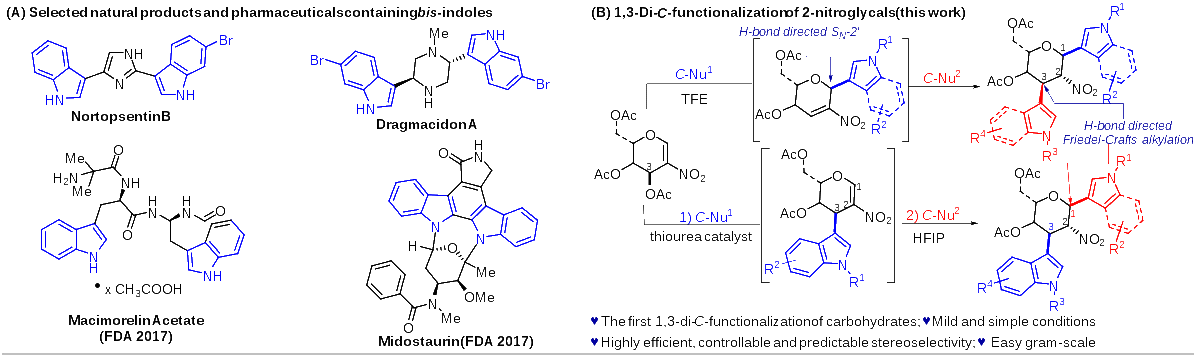
图1. 含双吲哚的活性分子以及C1,C3双吲哚糖衍生物的合成新方法
作者通过一系列条件筛选发现,以2,2,2-三氟乙醇(TFE)为溶剂和促进剂,在分子筛(MS)存在下,2-硝基稀糖和吲哚在室温下反应,以单一的非对映体形成了C1,C3-双吲哚化取代的糖衍生物3a,分离收率为92%。作者随后在该反应条件下,考察了不同取代吲哚和吡咯对全乙酰基硝基葡萄糖的反应性(图2):N-甲基保护的含有不同取代基的吲哚和吡咯以高收率和单一选择性得到产物,且吲哚取代基较为丰富,包括氯、溴、甲基、甲氧基、苄氧基和炔丙氧基等取代基;当以N-原子未保护的吲哚为原料时,该反应也可以顺利发生,但立体选择性有所降低,可能是由于TFE、C2-NO2和吲哚的N-H之间的多重氢键相互作用所致。
随后,作者考察了不同的2-硝基烯糖与吲哚的反应性:对于产物4a来说,该反应在克级规模时,也能顺利发生,产率略有降低但对选择性无明显影响;此外,作者也发现,通过C3-OAc的立体化学,可以预测产物中端基碳的立体化学,比如当以C3-位具有竖健(a-键)的OAc的硝基烯糖1a为原料时,产物3a中的端基碳也处于竖键的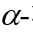 构型;相反,当以C3-位具有横键(e-键)的OAc的硝基烯糖1b为原料时,产物4a中的端基碳也处于横键的
构型;相反,当以C3-位具有横键(e-键)的OAc的硝基烯糖1b为原料时,产物4a中的端基碳也处于横键的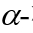 构型,而C2和C3位置的立体化学不受影响。其它常见乙酰化半乳糖硝基烯糖、乙酰化鼠李糖硝基烯糖等也均能以不错的收率和单一选择性得到相应产物,乳糖这类二糖的硝基烯糖也能在该条件下完成转化。
构型,而C2和C3位置的立体化学不受影响。其它常见乙酰化半乳糖硝基烯糖、乙酰化鼠李糖硝基烯糖等也均能以不错的收率和单一选择性得到相应产物,乳糖这类二糖的硝基烯糖也能在该条件下完成转化。
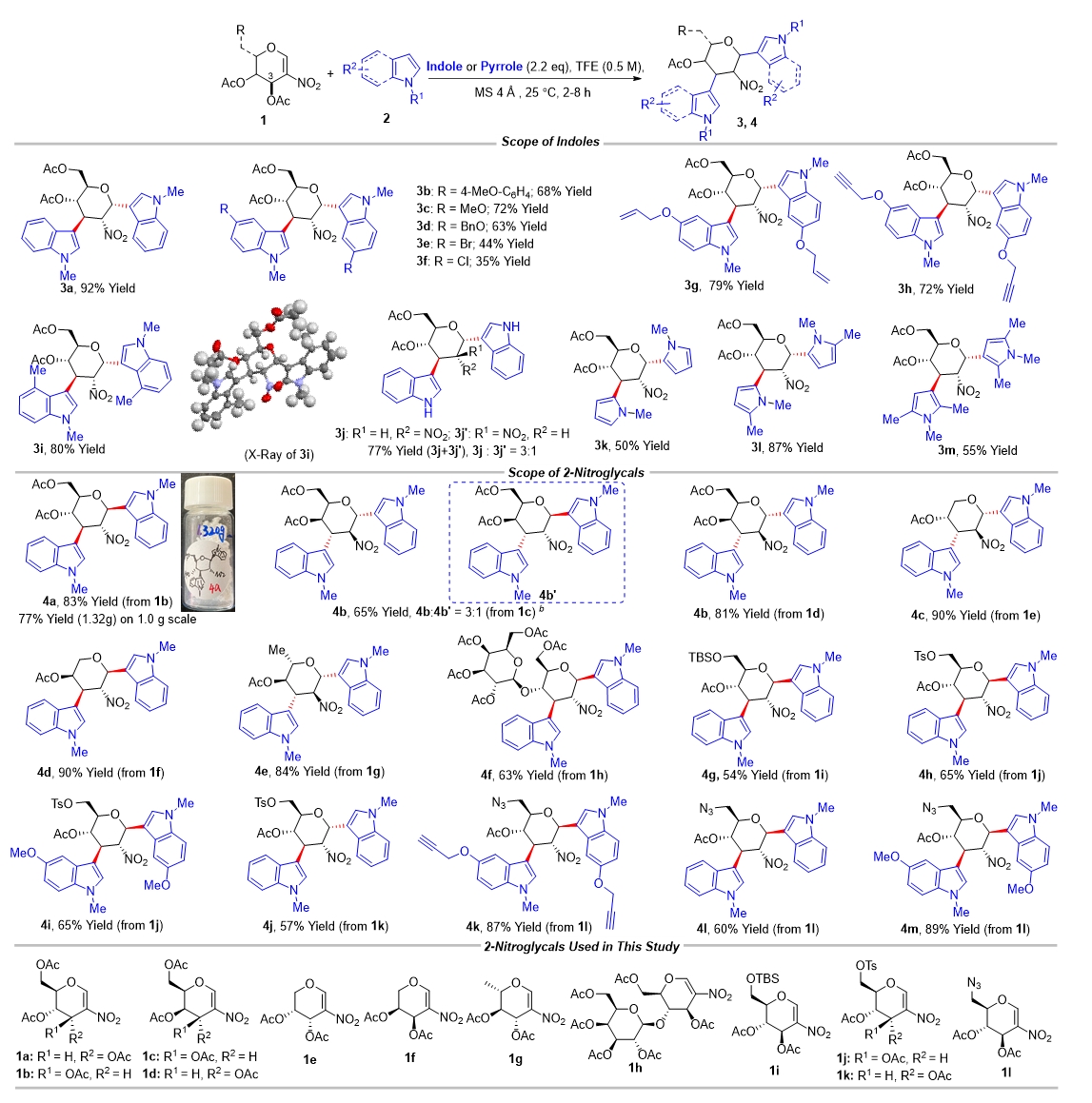
图2 2-硝基烯糖的C1,C3-双官能团化底物范围
作者在考察反应底物范围时发现,降低反应温度和减少吲哚的用量,可以分离得到Ferrier重排的2-硝基-吲哚-C-苷中间体,该中间体可以进一步在另一分子吲哚存在时,在室温到50 °C的反应温度下,发生Michael加成反应,形成C1和C3位置不同吲哚取代的糖衍生物。基于此策略,作者通过使用不同取代基的吲哚以及不同的硝基烯糖(除乙酰化的半乳糖硝基烯糖以外),采用两步一锅法,合成得到了系列C1和C3位置不同吲哚取代的糖衍生物,并获得了较高的产率、优秀的立体选择性和区域选择性,且可以以良好收率进行克级反应(图3)。
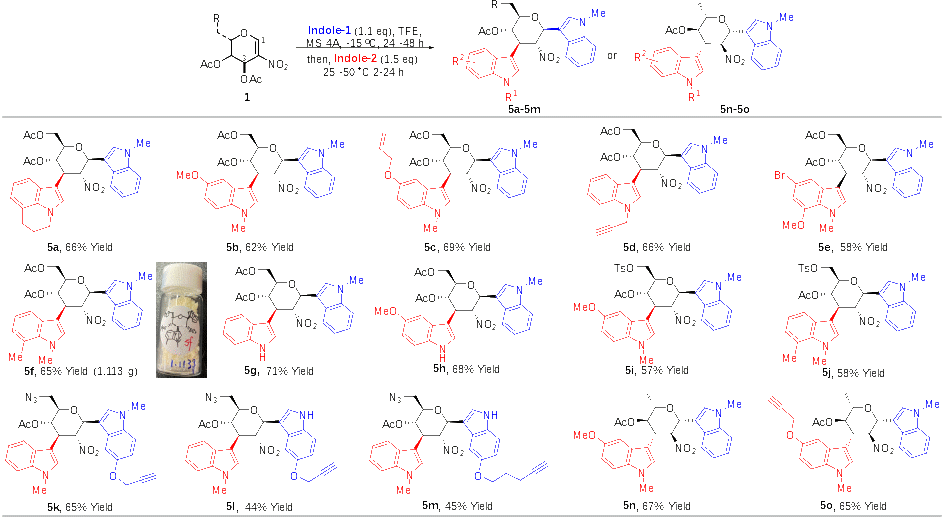
图3 C1和C3位置不同吲哚的C1,C3-双吲哚化糖苷的制备
由于作者无法通过上述图3的方法,高立体选择性地合成C1和C3位置不同吲哚取代的半乳糖衍生物,为了改变这一困境,作者通过优化反应条件,发现以双功能硫脲为催化剂时,可以以较高收率、高立体选择性和高区域选择性得到C3吲哚取代的2-硝基半乳糖烯糖。并且该反应底物范围广泛,不同位置单取代吲哚、多取代吲哚、吡咯均可以适用于该反应。为了比较,作者也发现,其它硝基烯糖也可以应用到该反应中,得到C3吲哚取代的2-硝基烯糖(图4)。
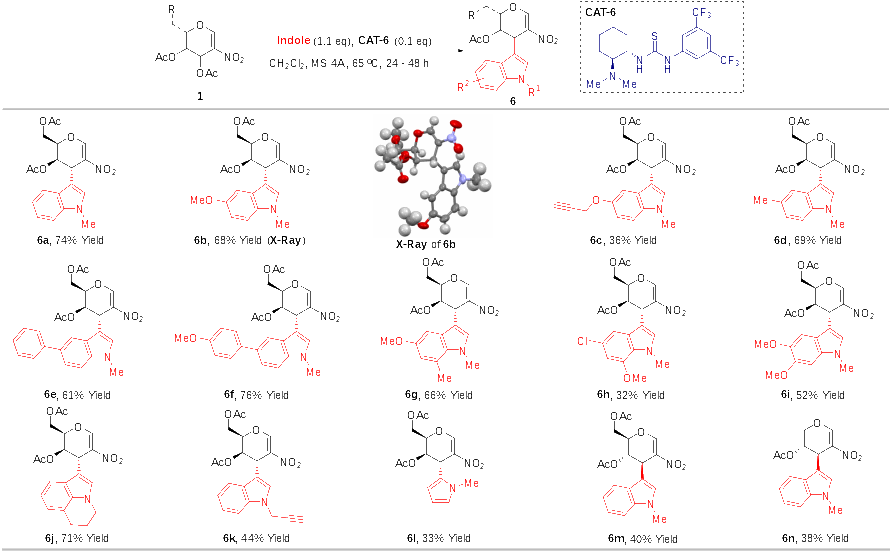
图4 3-吲哚-2硝基烯糖的制备
随后,作者以C3吲哚取代的2-硝基烯糖6为原料,以1,1,1,3,3,3-六氟异丙醇(HFIP)为溶剂,在4 Å分子筛存在的条件下,成功地使6与另一分子吲哚或吡咯反应,合成了系列C1,C3-双吲哚取代、C3-吲哚-C1-吡咯取代的半乳糖衍生物(图5)。作者发现,在合成C1,C3-双吲哚取代的半乳糖衍生物时,该反应非对映选择性较差(图4,4a,7b-7d),但是在合成C3-吲哚-C1-吡咯取代的半乳糖衍生物时(7a),该反应具有很高的非对映选择性,在其它糖的C3-吲哚-C2-硝基烯糖的底物中,作者发现了类似的现象。
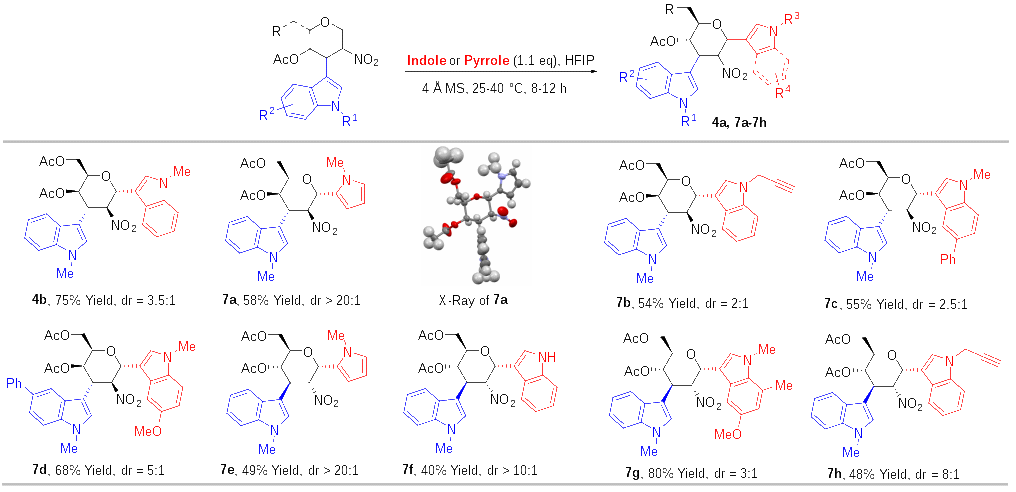
图5 从C3-吲哚硝基烯糖合成C1,C3-双取代糖衍生物
最后,作者也对该反应的产物进行了一些简单的衍生化(图6)。产物中的硝基,可以经雷尼Ni催化还原,并原位保护,转化为2-磺酰胺基吲哚糖衍生物(9),而乙酰基可以在碱存在下,高效地脱除形成糖衍生物10。同时,由于部分产物中,既含有N3基团,又含有炔基,因此,在Click反应条件下,作者成功地合成了系列糖桥环大环类化合物(11-13)。而且,C1-吲哚-C2-硝基烯糖,也可以在TFE中,与吡咯反应,高效、高立体选择性地合成C1-吲哚-C3-吡咯糖衍生物,进一步展现出了该方法在合成1,3-双-C-取代的糖衍生物方面的巨大潜力。最后,作者也对所获得的部分化合物进行了活性评估,结果表明,部分C1-吲哚-C2-硝基烯糖如8a,8b对HCT 116(人结肠癌细胞)的细胞毒活性IC50分别为1.586 μM 和 1.134 μM,对T24人膀胱癌细胞的细胞毒活性IC50 分别为2.949 μM和 3.548 μM,对AGS(人胃腺癌细胞) 的细胞毒活性IC50分别为0.695 μM 和 0.764 μM,同时,大环类化合物11表现出选择性地对MKN-45(人胃腺癌细胞)的细胞毒活性IC50为2.416 μM。
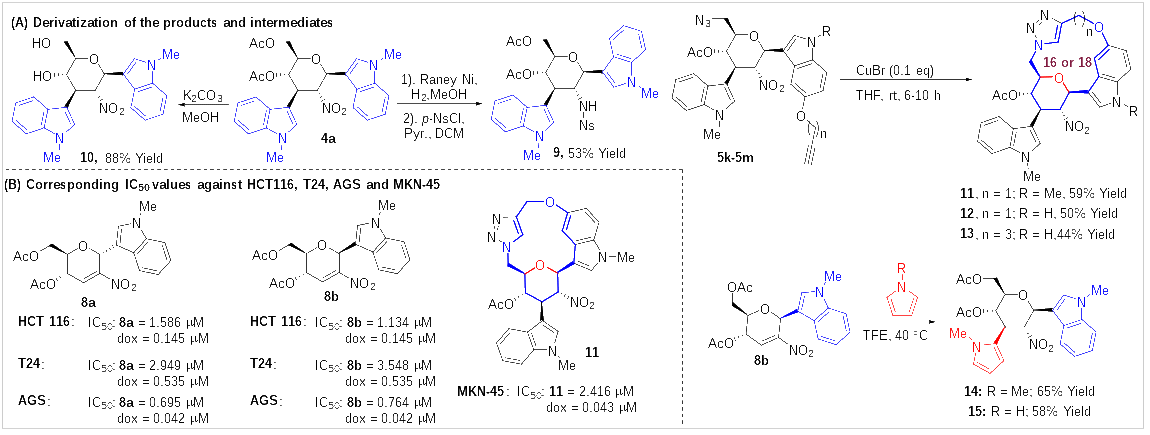
图6 产物的衍生化及部分化合物的活性评估
总之,中国科学院成都生物研究所马小锋研究员与其合作者报道了一种室温下,氟代醇溶剂(TFE)促进的、高效、高立体选择性的2-硝基糖的1,3-双-C-官能化反应。所需的硝基烯糖可以从烯糖经一步反应大规模制备,这为传统方法无法获得的 C1,C3-双-吲哚-、C1,C3-双吡咯-取代的糖衍生物提供了直接有效的合成方法。此外,通过C3位乙酰基的立体化学,可以预测反应产物中端基碳的α-或β-立体选择性;同时,通过对反应条件细微的改变,可以实现在糖的C1和C3位置上引入两种不同的吲哚;最后,当利用硫脲为催化剂时,能选择性地合成C3-吲哚取代-C2-硝基烯糖,其可以进一步在C1位发生官能团化修饰。结合这三种策略,作者高效、高立体选择性地制备了一系列具有挑战性的C1,C3-双-吲哚-、C1,C3-双吡咯-、C1-吲哚-C3吡咯-、C1-吡咯-C3吲哚-取代的糖衍生物。生物活性评价表明,3个化合物对T24、HCT116、AGS和MKN-45细胞具有较强的抗肿瘤活性,IC50在0.695 ~ 3.548 μM之间,而对人正常肝细胞(L-02)具有较低的细胞毒性,这将有利于高效、低毒的新型抗肿瘤化合物的进一步研发。
该研究以“Controllable 1,3-Bis-Functionalization of 2‑Nitroglycals with High Regioselectivity and Stereoselectivity Enabled by a H‑Bond Catalyst”为题发表于JACS Au(JACS Au2024,4,974−984)。中国科学院成都生物研究所马小锋研究员为本文的通讯作者,博士研究生李江涛为文章第一作者。同时,与该研究相关的内容已经申请中国发明专利四项(一种3-吲哚或吡咯基-2-硝基烯糖类化合物及其制备方法与应用,专利申请号:202410059158.X;一种2-硝基-2-烯吲哚糖碳苷类化合物及其合成方法与应用,专利申请号:202310766858.8,一种吲哚糖碳苷类大环化合物及其制备方法与应用,专利申请号:202310691698.5;吲哚糖苷类化合物及吡咯糖苷类化合物及合成方法与应用。专利申请号:202211581675.0)。上述研究得到国家自然科学基金(22377123、22001246)、四川省科技计划项目(2022ZYD0047、2022JDRC0132)、中国科学院生物资源计划(KFJBRP-008)的支持。
