




一个国际性的研究小组报告了普通小麦的完全注释版参考基因组;普通小麦是世界上最重要且种植面最广的农作物之一。这一具里程碑意义的基因组测序为加快研发旨在解决人类营养和提高作物恢复能力所需的小麦新品种而给科学家们提供了强有力的新工具。这一进展在两篇《科学》报告及一篇《科学进展》中进行了重点介绍,它们都强调了这一高品质基因组对了解小麦生物学的影响以及其对基因组辅助育种创新的潜力。
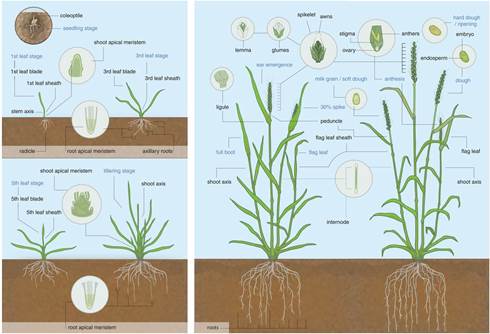
小麦是全球营养的一个主要来源;随着人口的快速增长及环境的不断变化,对小麦新品种的需求变得十分强劲,这些新品种应能提高产量、增加营养性、抗病虫害并能耐受生长条件的变化。详尽的作物基因组序列可通过对作物最有帮助的基础特性提供指导而给创建更强健作物品种带来重大进展;然而,现代的普通小麦是3种不同先祖基因组的复杂混合,因此,要充分解码其基因组一直颇具挑战性。
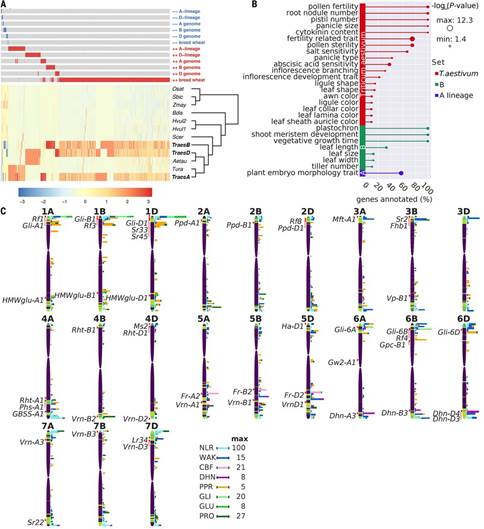
在一篇《科学》报道中,“国际小麦基因组测序协会”(IWGSC)报告了普通小麦品种“中国春”的第一个高品质完全注释参考基因组序列。“IWGSC参考序列”(IWGSC Reference Sequence,或RefSeqv1.0)宛如地图集,它提供了这一小麦品种的整个21条染色体的10万7000多个基因及超过400万个标记的位置和构建——它们中的某些与农业上重要的特性相关。据作者披露,被称作RefSeqv1.0的基因序列可以若干方式用于研究,其中包括基于CRISPR的基因组修饰。
在《科学》杂志中的第二则研究用这一新的参考基因组来进行同源基因(即功能类似但源于不同先祖基因组的基因拷贝)表达的全基因组分析。精确定位普通小麦中的这些同源基因可帮助科学家们更好地了解多倍体小麦的基础生物学。通过将基因表达数据集与IWGSC RefSeqv1小麦基因组序列结合,Ricardo Ramirez-Gonzalez等揭示了小麦中跨越不同组织、发育阶段及栽培品种的同源基因间的基因表达平衡。在发育时及在应激时,Ramirez-Gonzalez等在基因表达和共表达网络中发现了组织特异性偏差。据作者披露,他们的研究为小麦中具宝贵农业特性基因的设靶提供了框架结构。
最后,在《科学进展》(Science Advances)的一篇研究中,科研人员利用新的IWGSC参考序列对促成各种小麦引发的免疫性疾病及过敏的蛋白进行了仔细检查;这些疾病包括:乳糜泻、面包师哮喘及小麦依赖性运动诱发型过敏反应(WDEIA)等。在某些人中,接触小麦中的特定蛋白会引发严重的过敏反应。以乳糜泻为例,这种全球流行的慢性炎症性疾病是由醇溶蛋白(麦胶蛋白和麦谷蛋白)触发的,这两种蛋白在小麦中都是常见的。此外,呼吸系统或皮肤接触其它某些类型的小麦蛋白也与不良的免疫反应有关。然而,由于小麦基因组的复杂性及缺乏这一作物的完整基因组信息,人们仍然无法详细了解这些蛋白的性质。Angela Juhász等人用IWGSC RefSeqv1.0小麦基因组来搜寻那些编码已知的可诱发过敏的小麦蛋白基因,并对整个DNA序列中的每一该类基因进行了绘测。Juhász等人的分析确认了828种可能与免疫反应性蛋白有关的已知和先前未知的基因。结果显示,与乳糜泻和WDEIA有关的基因会在淀粉质胚乳组织(即烘焙面粉的来源)中表达,而各种脂质转移蛋白和α-淀粉酶胰蛋白酶抑制物基因族则与面包师哮喘有关。此外,本研究揭示,在开花期间的温度应激可增加主要的引发乳糜泻和WDEIA蛋白的表达。研究人员的详细分析就环境与生长条件对人类消费者造成麻烦的蛋白表达的作用提供了重要的线索。除了其它对食品工业有用的信息外,他们的研究还为低过敏性小麦品种的生产提供了资料。(来源:生物通)
Shifting the limits in wheat research and breeding using a fully annotated reference genome
Abstract An annotated reference sequence representing the hexaploid bread wheat genome in 21 pseudomolecules has been analyzed to identify the distribution and genomic context of coding and noncoding elements across the A, B, and D subgenomes. With an estimated coverage of 94% of the genome and containing 107,891 high-confidence gene models, this assembly enabled the discovery of tissue- and developmental stage–related coexpression networks by providing a transcriptome atlas representing major stages of wheat development. Dynamics of complex gene families involved in environmental adaptation and end-use quality were revealed at subgenome resolution and contextualized to known agronomic single-gene or quantitative trait loci. This community resource establishes the foundation for accelerating wheat research and application through improved understanding of wheat biology and genomics-assisted breeding.
原文链接:http://science.sciencemag.org/content/361/6403/eaar7191/tab-pdf
The transcriptional landscape of polyploid wheat
Abstract The coordinated expression of highly related homoeologous genes in polyploid species underlies the phenotypes of many of the world’s major crops. Here we combine extensive gene expression datasets to produce a comprehensive, genome-wide analysis of homoeolog expression patterns in hexaploid bread wheat. Bias in homoeolog expression varies between tissues, with ~30% of wheat homoeologs showing nonbalanced expression. We found expression asymmetries along wheat chromosomes, with homoeologs showing the largest inter-tissue, inter-cultivar, and coding sequence variation, most often located in high-recombination distal ends of chromosomes. These transcriptionally dynamic genes potentially represent the first steps toward neo- or subfunctionalization of wheat homoeologs. Coexpression networks reveal extensive coordination of homoeologs throughout development and, alongside a detailed expression atlas, provide a framework to target candidate genes underpinning agronomic traits in wheat.
原文链接:http://science.sciencemag.org/content/361/6403/eaar6089/tab-pdf
Genome mapping of seed-borne allergens and immunoresponsive proteins in wheat
Abstract Wheat is an important staple grain for humankind globally because of its end-use quality and nutritional properties and its adaptability to diverse climates. For a small proportion of the population, specific wheat proteins can trigger adverse immune responses and clinical manifestations such as celiac disease, wheat allergy, baker’s asthma, and wheat-dependent exercise-induced anaphylaxis (WDEIA). Establishing the content and distribution of the immunostimulatory regions in wheat has been hampered by the complexity of the wheat genome and the lack of complete genome sequence information. We provide novel insights into the wheat grain proteins based on a comprehensive analysis and annotation of the wheat prolamin Pfam clan grain proteins and other non-prolamin allergens implicated in these disorders using the new International Wheat Genome Sequencing Consortium bread wheat reference genome sequence, RefSeq v1.0. Celiac disease and WDEIA genes are primarily expressed in the starchy endosperm and show wide variation in protein- and transcript-level expression in response to temperature stress. Nonspecific lipid transfer proteins and α-amylase trypsin inhibitor gene families, implicated in baker’s asthma, are primarily expressed in the aleurone layer and transfer cells of grains and are more sensitive to cold temperature. The study establishes a new reference map for immunostimulatory wheat proteins and provides a fresh basis for selecting wheat lines and developing diagnostics for products with more favorable consumer attributes.
原文链接:http://advances.sciencemag.org/content/4/8/eaar8602/tab-pdf