




大麦是一种被广泛种植的谷物,通常用来制造啤酒和其他酒精性饮料,具有一个大而高度重复的基因组,很难完全测序。目前,由美国加州大学里弗赛德分校(UCR)的科学家带领的一个国际团队,从2000年开始进行的大麦基因组测序工作,达到了一个新的里程碑。研究人员对基因组中的绝大部分序列进行了测序,总共含有近三分之二的大麦基因。
相关研究结果发表在最近的《The Plant Journal》杂志,不仅将扩大遗传学家对于大麦DNA的知识面,也有助于我们在基因水平上理解小麦和其他食物来源。它还可以应用于植物育种,增加某些特性(如麦芽品质或杆锈)标记的精确度。延伸阅读:我科学家参与完成非洲稻基因组测序。
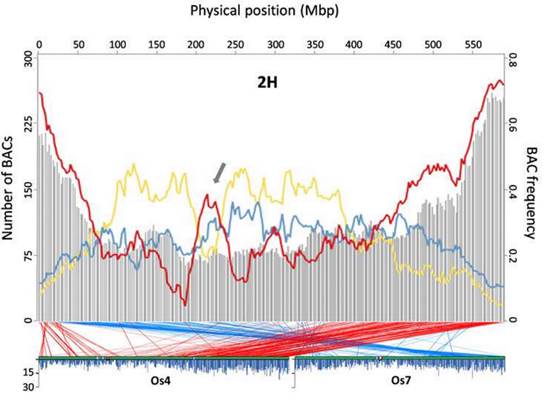
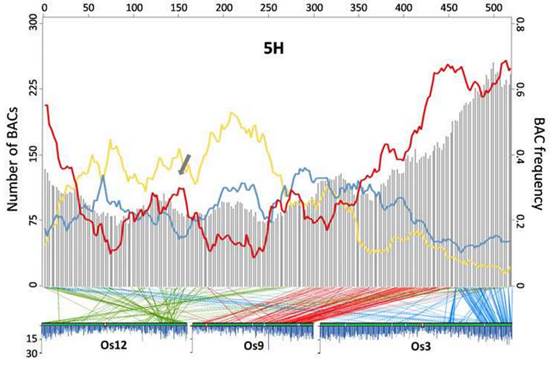
本研究论文的通讯作者、加州大学里弗赛德分校遗传学教授Timothy J. Close说:“我们现在对大麦基因组中的遗传信息有了更高的分辨率。这是世界各地使用的一种改良资源。在这项工作之前,一个长期持有的观点是,基因在大麦、小麦及其近缘种基因组中的分布是这样的,基因密集区域只是在染色体的末端附近,那里也有一个很高的重组率。我们的工作揭示了明确的例外,确定了基因丰富、但低重组的不正常地区。”
重组指的是,在减数分裂过程中自然形成新的基因组合,这是细胞周期中的一个阶段,在这个阶段染色体配对并进行交换。Close解释说,植物育种者依赖于减数分裂重组,把麦芽品质、杆锈或许多性状相关基因的有利形式,引入到栽培品种中。杂交并对后代植物进行了筛选,寻找理想的新特性组合。当一个基因的有利形式(等位基因)位于一个基因密集低重组区域时,需要开展更多的工作,将有利的等位基因引入现有的品种,而不会牵涉邻近的基因——可能存在于不良的形式。
Close说:“例如,育种者可能成功地加入了一个来自于野生大麦的、有利于茎锈病的、良好的等位基因,但随着这个基因转移的还有另一个基因,从而导致成熟麦穗的脱落。现在,育种学家将会得到一种茎锈病抗性的植物,但是在收获之前,种子都掉到地上,而不是留在植物上。所以,如果一个基因位于一个基因密集的重组区域,那么这就意味着,杂交后必须对更大数量的后代进行检测,以寻找那些来自罕见重组事件的后代,将所需的新等位基因与邻近基因的不良形态分离开来。了解基因密集的低重组区域的位置,有助于决定哪些基因有助于品种改良。”
Close与UCR计算机科学和工程教授Stefano Lonardi联手,制定了一套有效的计算创新算法,帮助进一步测定大麦基因组序列。这种新的算法可以处理更大型的数据集,使研究人员能够比其他可能的方法获得更多的进展。
Lonardi说:“Tim和我能够在项目所有的步骤中紧密合作,从湿实验室的实验设计,到最终的结果分析;面临的主要挑战是,如何处理非常大的大麦样品数量,为此我们设计了一种新型的测序方法,利用组合数学中的深结果。”
Close解释说:“当育种学家们进行杂交以开发新品种时,他们想寻找更适合于农业环境或作物产品市场的新基因组合。这通常意味着,与一些远缘个体进行杂交,它们可能会在基因组中的许多位置携带不利的等位基因。当育种学家能够只杂交有利的等位基因时,那将是有益的。”
他补充说:“如果邻近基因的不利等位基因始终与靶基因的良好等位基因共同出现,那么这就不是有益的。当一个有利的等位基因位于一个区域内,在那里重组很少打破与邻居基因坏等位基因的连锁,育种学家将不得不更努力地工作,在一个更大的种群中找到合适的组合。通过找出大麦基因组的哪个区域抗重组,育种学家将能够做出更明智的决策。”
大约20年前,Close几乎完全从事于脱水蛋白(dehydrins)——一个蛋白家族,所有植物用以响应干旱胁迫或低温。到2000年,他的研究小组了解到,大麦中至少有13个脱水蛋白基因,Close想研究它们。
他说:“大麦的基因组资源是不充分的,幸运的是,我经过适当的培训,可帮助开发基本的基因组资源。我和UCR的同事们已经完成了最后的基因组资源任务,仍然还需要很多时间来研究大麦脱水素基因。”
为了在高分辨率上获得大麦基因组的含基因部分,Close和他的团队鉴定并测定了15,622个BAC或细菌人工染色体——大麦DNA的小片段与其他DNA连接以构成一个环状分子,可以在大肠杆菌细菌的细胞内复制和传播,从而使研究人员能够产生每个BAC的副本,用于大麦基因组一小部分的DNA测序。通过探索这些测定的BAC(包含大约三分之二的大麦基因),Close和他的团队发现,基因丰富的区域,不仅仅存在于高组合的区域。
Close说:“在低重组区域,有基因丰富的区域,这对于植物育种是非常重要的。”因为大麦与小麦有着密切的亲缘关系,这项新的工作可以提供有用的信息,促进小麦基因组的完全测序。(来源:生物通 王英)
Sequencing of 15,622 gene-bearing BACs clarifies the gene-dense regions of the barley genome
Abstract Barley (Hordeum vulgare L.) possesses a large and highly repetitive genome of 5.1 Gb that has hindered the development of a complete sequence. In 2012, the International Barley Sequencing Consortium released a resource integrating whole-genome shotgun sequences with a physical and genetic framework. However, because only 6,278 BACs in the physical map were sequenced, fine structure was limited. To gain access to the gene-containing portion of the barley genome at high resolution, we identified and sequenced 15,622 BACs representing the minimal tiling path of 72,052 physical-mapped gene-bearing BACs. This generated ~1.7 Gb of genomic sequence containing an estimated 2/3 of all Morex barley genes. Exploration of these sequenced BACs revealed that although distal ends of chromosomes contain most of the gene-enriched BACs and are characterized by high recombination rates, there are also gene-dense regions with suppressed recombination. We made use of published map-anchored sequence data from Aegilops tauschii to develop a synteny viewer between barley and the ancestor of the wheat D genome. Except for some notable inversions, there is a high level of collinearity between the two species. The software HarvEST:Barley provides facile access to BAC sequences and their annotations, along with the barley-Ae. tauschii synteny viewer. These BAC sequences constitute a resource to improve the efficiency of marker development, map-based cloning, and comparative genomics in barley and related crops. Additional knowledge about regions of the barley genome that are gene-dense but low-recombination is particularly relevant.
原文链接:http://onlinelibrary.wiley.com/doi/10.1111/tpj.12959/epdf